
















Scientists have developed a powerful new computational framework, PathGennie, that promises to significantly accelerate the simulation of rare molecular events—processes that are essential to drug development but typically require enormous computational resources.
The breakthrough, recently published in the Journal of Chemical Theory and Computation, offers a major advance for computer‑aided drug discovery by enabling accurate prediction of how drug molecules detach from their protein targets without introducing artificial distortions.
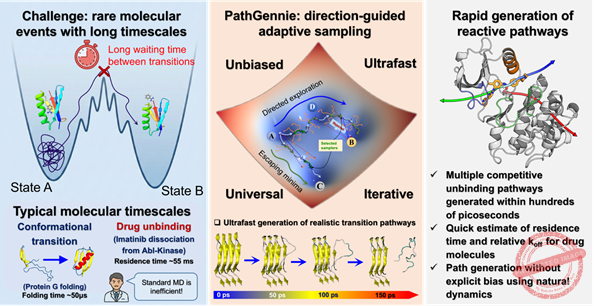
In modern drug development, understanding a drug’s residence time—how long it remains bound to its target protein—is often more important than its binding strength.
However, capturing the unbinding process is extremely challenging because such events unfold over milliseconds to seconds, far beyond the reach of classical molecular dynamics (MD) simulations, even on state-of-the-art supercomputers. To overcome this, researchers traditionally apply artificial forces or raise temperatures to hasten the event, but these methods can alter the true physical behavior of the system.
Developed by researchers at the S. N. Bose National Center for Basic Sciences, Kolkata, an autonomous institute under the Department of Science and Technology (DST), PathGennie takes a fundamentally different and distortion‑free approach. Instead of forcing molecular motion, the algorithm mimics a natural selection process at the microscopic scale.
It generates large numbers of ultrashort, unbiased MD trajectories—each lasting just a few femtoseconds—and selectively extends only those that show genuine progress toward the unbinding event.
This strategy functions like a guided “scouting mission” through a molecule’s energy landscape. Productive fragments are continued, while unproductive ones are discarded. The “survival of the fittest” approach enables PathGennie to bypass long waiting times without applying any biasing force or elevated temperature, ensuring that the resulting transition pathways remain physically accurate. The general purpose framework can operate in any set of collective variables (CVs), including high‑dimensional or machine‑learned feature spaces, making it compatible with modern simulation workflows.
In proof‑of‑concept demonstrations, the team led by Prof. Suman Chakrabarty, along with Dibyendu Maity and Shaheerah Shahid, showed that PathGennie could identify multiple competing pathways for challenging systems.
It mapped how a benzene molecule escapes the deep binding pocket of the T4 lysozyme enzyme, revealing a network of distinct ligand‑exit routes. The tool also identified three separate dissociation pathways for the anti‑cancer drug imatinib (Gleevec) unbinding from Abl kinase—recovering all previously reported routes in just a few iterations. These pathways were identified without any external bias, yet matched observations from biased simulations and experimental results.
According to the researchers, PathGennie’s versatility extends beyond drug–target interactions. It can be applied to chemical reactions, catalytic processes, phase transitions, self‑assembly, and other systems involving transitions













{kind=link}